Research Accomplishments
Overview
During my research at the University of Illinois,I focused on three primary areas of research.
- Superfluid hydrodynamics,quantum turbulence and the dynamics of topological defects
- Developing bioinformatics pipelines for state-of-the-art metagenomic surveys of environmental DNA
- Exploring the evolutionary dynamics of gastrointestinal microbiomes
Bioinformatics
Sequence-based analyses provide a very important tool set needed to understand the microbial environment on Earth. The design of these tools is challenging because they have to provide both a way to manipulate an increasing volume of sequence data coming from the current generation of sequencing platforms, and the ability to process and extract meaningful biological information to gain further insight on the ecological,evolutionary and environmental dynamics of microbiomes.
My work in this area has resulted in three projects. First, the creation of a pipeline for processing data from 16S rRNA hypervariable tag regions coming from high-throughput pyrosequencing platforms1, available for public use at the Tornado website. These sequences have traditionally been challenging to process due to the sheer size of these datasets,and thus making difficult to align and remove artifacts. The pipeline in this work takes as input two alignments of the same sequence set, one from the Ribosomal Database Project, and the other coming from the NAST aligner, either from the Greengenes website or the implementation of the aligner in the software package Mothur. The pipeline merges both alignments, creating a third alignment which is superior to both inputs, and makes detection and removal of artifacts an easier task. The pipeline also provides a linkage-based clustering tool to create Operational Taxonomic Units (OTU) from the dataset.
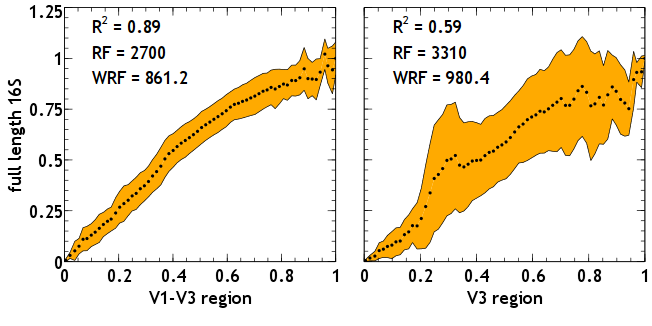
Comparison of trees from two 16S regions to the tree created using the full length gene. The longer V1-V3 region (left) is better suited than the V3 region alone (right).
Second, these sequence datasets, despite the massive sizes, only contain a short region of the 16S rRNA, and thus likely introduce artifacts when compared to datasets containing full-length 16S reads. There has been interest in the extent of these artifacts and how they affect metrics relevant to microbial ecology. Nonetheless, little is known about the kind of phylogenetic artifacts these short reads insert. My work focused on characterizing these phylogenetic artifacts, and how they could affect their suitability for environmental surveys of microbial populations2. I created datasets from randomly selected sequences from the Greengenes database, and I also created artificial short read libraries from these sequences. Then, for each library, I created phylogenetic trees and then compared the differences between the trees. I used several metrics for the comparison: a tip-to-tip distance between species in the trees and how it varies in each tree; the Robinson-Foulds (RF) distance between trees, which counts the number of different subtrees in a given pair of trees; and a weighted RF distance, which weights each different subtree by its support value. The results show that indeed there are substantial phylogenetic differences if short reads are used (for example V3 or V6) instead of the full-length reads, and these differences decrease as the length of the reads increases (for example reads containing regions V1 to V3). These results suggest that any metric derived from phylogenetic trees made using these short reads should be taken with skepticism, but the outlook looks good as the length of these reads increases.
Third and finally, I applied these knowledge on a problem of importance to microbial ecology and evolution. This problem revolves around the question of what kind of evolutionary dynamics are present in microbial environments, namely a niche-based evolution or a purely neutral evolution scheme,and is it possible to answer such a question from sequence data alone. We took 16S rRNA sequence data from gastrointestinal microbiomes from three mammals,and generated rank-abundance curves for them. Rank-abundance curves are ordered lists of abundance per OTU,sorted from high to low abundance. Other tests performed were clustering statistics of the datasets versus artificial statistics created from models of neutral and niche evolution. Our data shows features present both in niche (for high abundance organisms) and neutral (for low abundance organisms) evolution models,making it so far impossible to attribute a single evolution model to these communities, and instead open the possibility of the idea of a mixed system, where neutrally-evolved microbes take over a new niche whenever there is a change in their environment.
Efficient Algorithms for Simulation of Quantum Fluids
Simulation of quantum vortices in a complex flow.
Quantum fluids, such as superfluid He-4, Bose-Einstein condensates (BEC) and the more exotic cores of neutron stars, have been of high interest because they have been ideal test beds to probe questions regarding the basic interactions of matter at low temperature. In particular, the hydrodynamics of quantum fluids possess unusual features when compared to normal fluids, such as the presence of vortices of quantized circulation and having no viscosity. These features have led to many questions on the consequences of such features, such as what is the nature of a turbulent quantum fluid? What features of classical turbulence are also seen in quantum turbulence? What are the effects of quantization of circulation in a turbulent quantum fluid? How can a turbulent quantum fluid dissipate energy when, ideally, the fluid has no viscosity? Besides experiment and theory, numerical simulation has played an important role in the study of turbulent fluids. Simulations have provided key information about the onset of turbulence, and the behavior of fluids near rough boundaries, due to being able to provide high resolution data of the motion of the fluid in these situations. Due to the multi-scale and non-linear nature of turbulence, direct numerical simulation of fluids is highly demanding of computational resources, and even more so as the need for detail increases. In the case of quantum fluids, the problem is worse, as models used to properly describe these fluids rely on not one but two fluid fields, increasing the complexity of the simulation problem. Simplifications have been made by, for example, simulating only the dynamics of the quantized vortices, but these simulations do not scale well as the density of vortices is increased.
I created an efficient model for quantum fluids based on the Ginzburg-Pitaevskii equations for superfluids near the λ-transition point, using a Cell Dynamical System (CDS) approach. CDS models are a version of coupled map lattices that directly implement the symmetries and interactions of the field on a map, bypassing the necessity for a differential equation approach, allowing for much larger time steps compared to discretizations of differential equations. Even more, CDS allows for the creation of models whose equivalent differential equation formulation does not exist. CDS models have been created for systems ranging from simple two-phase systems, to uniaxial and biaxial liquid crystals, to modeling of travertine terraces at Yellowstone National Park accounting only for the fluid dynamics and deposition chemistry of calcium carbonate.
Using this CDS model for quantum fluids I was able to obtain the probability density of the speed of the topological defects,and found that the high-speed tail of the distribution decays as power law with exponent -3, not only in 2 dimensions as predicted elsewhere, but also in 3 dimensions, and that this distribution shows Gaussian correction due to the finite size of the defect cores3. This result applies to quantum vortices, crystal dislocations and, in principle, any topological defects whose interaction decay as 1/r, with r being the distance between defects.
In short, this is the first simulation of superfluids that can, at the same time, handle large system sizes and large numbers of vortex lines without using a vortex filament formalism or a local induction approximation, allowing reconnections and vortex-phonon interactions to be captured in a computationally efficient way.
-
M. Sipos, P. Jeraldo, N. Chia, A. Qu, A.S. Dhillon, et al., Robust Computational Analysis of rRNA Hypervariable Tag Datasets, PLoS ONE 5, e15220, (2010) ↩
-
P. Jeraldo, N. Chia and N. Goldenfeld, On the Suitability of Short Reads of 16S rRNA for Phylogeny-based Analyses in Environmental Surveys, Environ. Microbiol. 13, 3000-3009 (2011)↩
-
L. Angheluta, P. Jeraldo, K. Dahmen and N. Goldenfeld, Avalanche statistics and intermittency in topological defect-dominated flows, in review, arXiv:1103.2185↩